Un modèle d’intelligence artificielle trouve des molécules médicamenteuses potentielles mille fois plus rapidement
Un modèle géométrique d’apprentissage en profondeur est plus rapide et plus précis que les modèles informatiques de pointe, ce qui réduit les risques et les coûts d’échec des essais de médicaments.
L’intégralité de l’univers connu regorge d’un nombre infini de molécules. Mais quelle fraction de ces molécules possède des caractéristiques médicamenteuses potentielles qui peuvent être utilisées pour développer des traitements médicamenteux salvateurs ? Des millions? Des milliards ? Des billions ? La réponse : novemdecillion, ou 1060. Ce nombre gargantuesque prolonge le processus de développement de médicaments pour les maladies à propagation rapide comme le Covid-19, car il va bien au-delà de ce que les modèles de conception de médicaments existants peuvent calculer. Pour mettre les choses en perspective, la Voie lactée compte environ 100 milliards, ou 1011 étoiles.
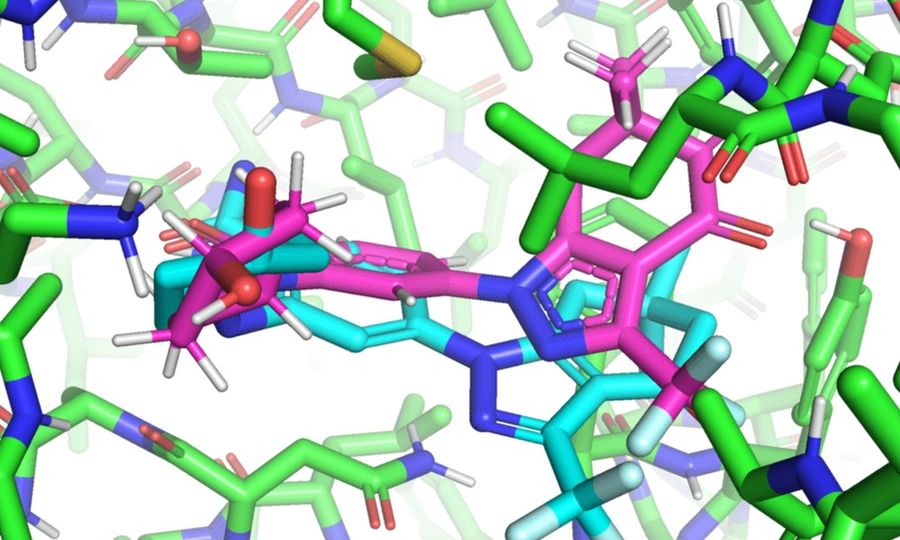
Dans un article qui sera présenté à la Conférence internationale sur l’apprentissage automatique (ICML), les chercheurs du MIT ont développé un modèle géométrique d’apprentissage en profondeur appelé EquiBind qui est 1 200 fois plus rapide que l’un des modèles d’amarrage moléculaire computationnel les plus rapides, QuickVina2-W, dans lier avec succès des molécules de type médicament à des protéines. EquiBind est basé sur son prédécesseur, EquiDock, qui se spécialise dans la liaison de deux protéines à l’aide d’une technique développée par feu Octavian-Eugen Ganea, un récent laboratoire d’informatique et d’intelligence artificielle du MIT et la clinique Abdul Latif Jameel pour l’apprentissage automatique en santé (Clinique Jameel) postdoc, qui a également co-écrit l’article EquiBind.
Avant même que le développement de médicaments puisse avoir lieu, les chercheurs en médicaments doivent trouver des molécules prometteuses de type médicament qui peuvent se lier ou «s’ancrer» correctement sur certaines cibles protéiques dans un processus connu sous le nom de découverte de médicaments. Après avoir réussi à s’arrimer à la protéine, le médicament de liaison, également connu sous le nom de ligand, peut empêcher une protéine de fonctionner. Si cela arrive à une protéine essentielle d’une bactérie, cela peut tuer la bactérie, conférant une protection au corps humain.
Cependant, le processus de découverte de médicaments peut être coûteux à la fois financièrement et informatiquement, avec des milliards de dollars investis dans le processus et plus d’une décennie de développement et de tests avant l’approbation finale de la Food and Drug Administration. De plus, 90 % de tous les médicaments échouent une fois qu’ils sont testés sur des humains en raison de l’absence d’effets ou de trop d’effets secondaires. L’une des façons dont les compagnies pharmaceutiques récupèrent les coûts de ces échecs est d’augmenter les prix des médicaments qui réussissent.
Le processus informatique actuel pour trouver des molécules candidates prometteuses pour les médicaments se présente comme suit : la plupart des modèles informatiques de pointe reposent sur un échantillonnage intensif de candidats couplé à des méthodes telles que la notation, le classement et le réglage fin pour obtenir le meilleur « ajustement » entre le ligand et la protéine.
Hannes Stärk, auteur principal de l’article et étudiant diplômé de première année conseillé par Regina Barzilay et Tommi Jaakkola du département de génie électrique et d’informatique du MIT, compare les méthodologies typiques de liaison ligand-protéine à “essayer d’insérer une clé dans une serrure avec beaucoup de trous de serrure. Les modèles typiques prennent beaucoup de temps pour noter chaque « ajustement » avant de choisir le meilleur. En revanche, EquiBind prédit directement l’emplacement précis de la clé en une seule étape sans connaissance préalable de la poche cible de la protéine, connue sous le nom de “blind docking”.
Contrairement à la plupart des modèles qui nécessitent plusieurs tentatives pour trouver une position favorable pour le ligand dans la protéine, EquiBind a déjà un raisonnement géométrique intégré qui aide le modèle à apprendre la physique sous-jacente des molécules et à généraliser avec succès pour faire de meilleures prédictions lorsqu’il rencontre de nouvelles données invisibles. .
La publication de ces résultats a rapidement attiré l’attention des professionnels de l’industrie, dont Pat Walters, responsable des données chez Relay Therapeutics. Walters a suggéré que l’équipe essaie son modèle sur un médicament et une protéine déjà existants utilisés pour le cancer du poumon, la leucémie et les tumeurs gastro-intestinales. Alors que la plupart des méthodes d’amarrage traditionnelles n’ont pas réussi à lier les ligands qui fonctionnaient sur ces protéines, EquiBind a réussi.
“EquiBind fournit une solution unique au problème d’amarrage qui intègre à la fois la prédiction de la pose et l’identification du site de liaison”, déclare Walters. “Cette approche, qui exploite les informations de milliers de structures cristallines accessibles au public, a le potentiel d’avoir un impact nouveau sur le domaine.”
“Nous avons été étonnés de constater que, alors que toutes les autres méthodes se sont complètement trompées ou n’en ont obtenu qu’une seule correcte, EquiBind a pu la mettre dans la bonne poche, nous avons donc été très heureux de voir les résultats pour cela”, déclare Stärk.
Bien qu’EquiBind ait reçu de nombreux commentaires de professionnels de l’industrie qui ont aidé l’équipe à envisager des utilisations pratiques du modèle informatique, Stärk espère trouver différentes perspectives lors de la prochaine ICML en juillet.
“Les commentaires que j’attends le plus avec impatience sont des suggestions sur la façon d’améliorer encore le modèle”, dit-il. “Je veux discuter avec ces chercheurs… pour leur dire ce que je pense être les prochaines étapes et les encourager à aller de l’avant et à utiliser le modèle pour leurs propres articles et pour leurs propres méthodes… nous avons déjà eu de nombreux chercheurs qui nous ont contactés et demander si nous pensons que le modèle pourrait être utile pour leur problème.
Ce travail a été financé, en partie, par le consortium Pharmaceutical Discovery and Synthesis; la clinique Jameel ; le programme DTRA Découverte des contre-mesures médicales contre les menaces nouvelles et émergentes ; le programme DARPA Accelerated Molecular Discovery; la bourse MIT-Takeda ; et la subvention NSF Expeditions Recherche collaborative : Comprendre le monde à travers le code.
Ce travail est dédié à la mémoire d’Octavian-Eugen Ganea, qui a apporté des contributions cruciales à la recherche sur l’apprentissage automatique géométrique et a généreusement encadré de nombreux étudiants – un brillant universitaire à l’âme humble.
.